





Ожирение в современном цивилизованном обществе приняло характер эпидемии. Всё больше и больше людей страдают от недостатка физической нагрузки и избыточного веса. Среди больных с ожирением отдельную группу составляют дети и подростки. Причины развития заболевания в этом случае заключаются не только в малоподвижном образе жизни и несбалансированном питании, но и в генетических аномалиях. К подобным патологиям относится синдром Прадера — Вилли.
Содержание
Наследственное ожирение: определение и вероятность развития
Вся информация о внешности человека, характере обменных процессов в организме заложена в молекуле дезоксирибонуклеиновой кислоты (ДНК). Участки ДНК, кодирующие определённые признаки (цвет волос, глаз, кожи), носят название генов. Весь наследственный материал человека сгруппирован в сорок шесть хромосом, отлично видимых в ядре клетки через микроскоп. Половину из них будущий ребёнок получает от отца, другую — от матери. Хромосомы первоначально содержатся в половых клетках — яйцеклетке и сперматозоиде.
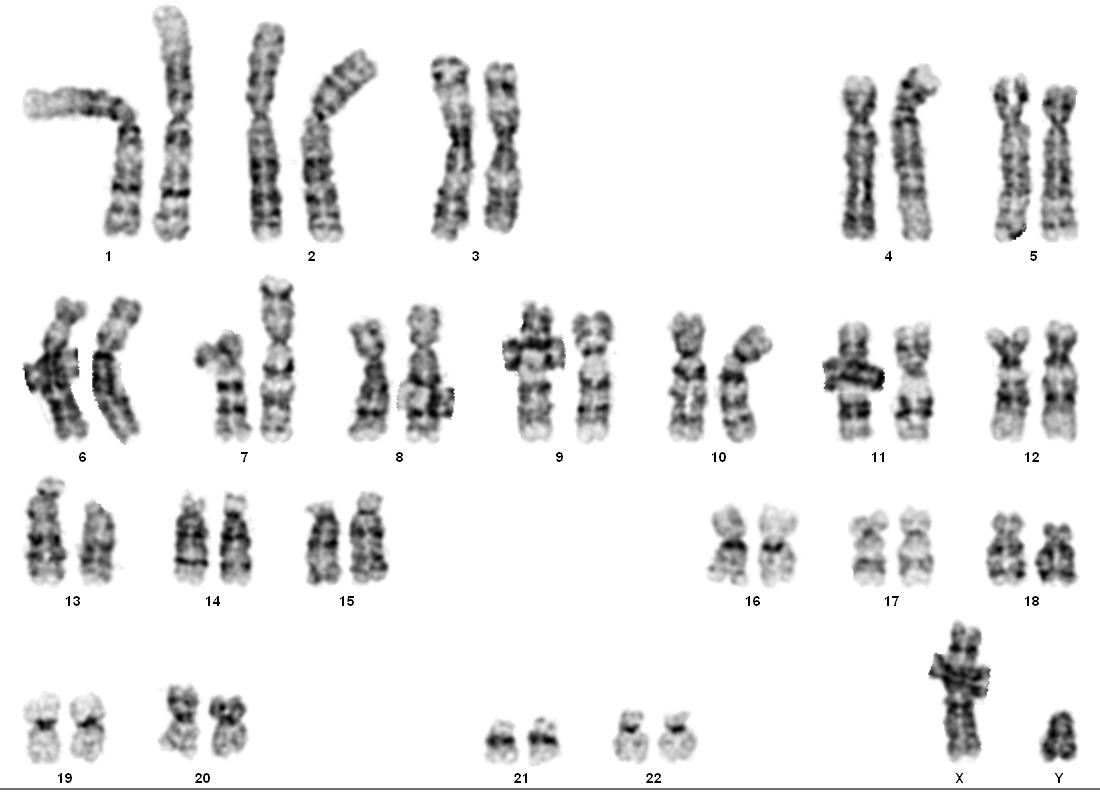
Хромосомный набор человека содержит всю информацию о строении и функционировании организма
Любое повреждение молекулы ДНК различной протяжённости (ген, участок хромосомы) с большой вероятностью скажется или на внешних чертах человека, или на характере обмена веществ. Синдром Прадера — Вилли — медицинский термин, описывающий характерные изменения внешнего вида больного в сочетании с ожирением наследственной природы и особенностями обмена веществ.

Синдром Прадера — Вилли — совокупность ожирения, изменённых черт лица и пропорций тела наследственной природы
Впервые синдром был описан учёными Андреасом Прадером и Генрихом Вилли в 1956 году. Распространённость патологии составляет один случай на десять — двадцать тысяч новорождённых. Заболевание одинаково часто встречается среди мальчиков и девочек. В литературе описаны семейные случаи синдрома Прадера — Вилли.
Наследственность как основная причина заболевания
Основная причина развития синдрома Прадера — Вилли — нарушение в структуре генов, расположенных в пятнадцатой хромосоме. К формированию болезни приводят два типа дефекта:
- потеря большого участка молекулы ДНК (микроделеция) отцовского происхождения;
- наследование обеих пятнадцатых хромосом от матери (изодисомия).
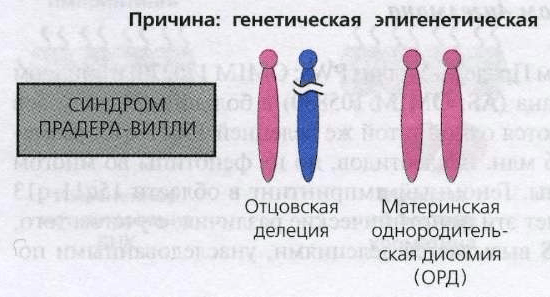
Синдром Прадера — Вилли развивается вслествие генетических дефектов пятнадцатой хромосомы
Около двух третей случаев патологии обусловлены микроделецией, остальные — материнской изодисомией.
Существует генетический противоположный дефект: микроделеция материнского происхождения или отцовская дисомия. Обе причины приводят к развитию наследственной патологии под названием синдром Ангельмана. Клиническая картина при этом заболевании характеризуется замедлением физического и интеллектуального развития, нарушением координации движений, эпилептическими судорожными приступами.
До развития молекулярной генетики было принято считать, что наследование обеих пар хромосом от одного родителя невозможно. Однако с появлением современных методов анализа был не только доказан этот факт. В процессе проводимых генетических исследований было опровергнуто утверждение о равноценном влиянии отцовской и материнской наследственной информации на внешние черты и характер обмена веществ ребёнка.
Клинические аспекты генетических заболеваний — видео
Особенности патологии
Основным следствием генетических аномалий при синдроме Прадера — Вилли является необычный обмен жиров в организме, приводящий к десятикратному превалированию их отложения в подкожно-жировой клетчатке над расщеплением. Ещё одним важным механизмом развития заболевания является нарушение обмена половых гормонов, в результате которого репродуктивная система имеет множество аномалий анатомического строения.
Кроме того, при синдроме Прадера — Вилли значительно повышен риск образования злокачественных опухолей вследствие генетически запрограммированной слабой системы защиты молекулы ДНК от поломок.
Симптомы заболевания
Клиническая картина при синдроме Прадера — Вилли у детей разного возраста существенно различается.
Признаки синдрома Прадера — Вилли в различных возрастных группах — таблица
| Возрастные группы | Новорождённые | 12–18 месяцев | Дошкольники и подростки |
| Показатели | |||
| Физическое развитие |
|
Выраженное снижение мышечного тонуса |
|
| Нервно-психическое развитие | Соответствует возрасту | Ослабление рефлексов | Задержка интеллектуального развития |
| Половое развитие | Соответствует возрасту, возможно отсутствие яичек в мошонке (крипторхизм) | Соответствует возрасту, возможно отсутствие яичек в мошонке (крипторхизм) | Недоразвитие половых органов, крипторхизм |
| Анатомические аномалии | Незначительно увеличены размеры черепа | Незначительно увеличены размеры черепа |
|
В период внутриутробного развития плод с синдромом Прадера — Вилли имеет незначительные анатомические особенности в виде узкого лицевого скелета.
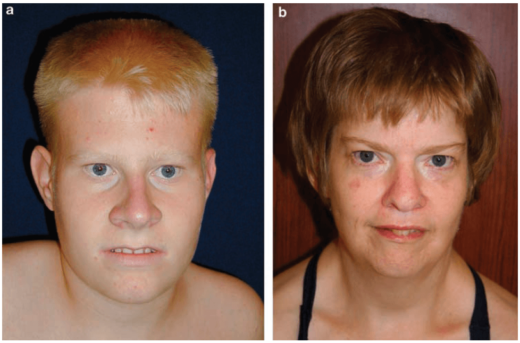
Внешний вид больных с синдромом Прадера — Вилли — фото
-

- Генетический дефект пятнадцатой хромосомы приводит к множественным внешним проявлениям болезни
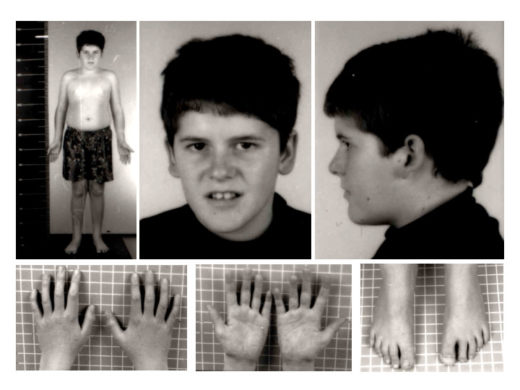
-

- Ожирение при синдроме Прадера — Вилли в основном затрагивает туловище, плечи и бёдра
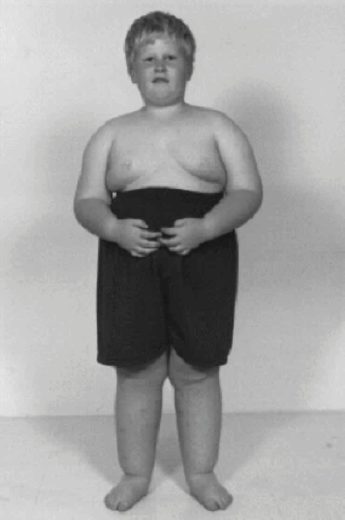
-

- Ожирение — самая характерная внешняя черта заболевания

-

- Широкая переносица, узкие глаза и губы — характерные черты лица больных синдромом Прадера — Вилли
Методы диагностики
Для установления диагноза используются следующие методики:
- тщательный осмотр врача — обязательно с выяснением всех деталей заболевания, включая особенности периода беременности, родов и ранних этапов развития ребёнка;
- измерение роста и веса, длины сегментов конечностей — проводится с целью определения важнейших признаков болезни: избыточного веса, низкого роста, диспропорции конечностей;

Диспропорция конечностей — один из основных внешних признаков синдрома Прадера — Вилли
- неврологический осмотр — для грудного ребёнка позволяет выявить ослабление рефлексов и снижение мышечного тонуса. В более старшем возрасте осмотр преследует цель определить степень нарушения интеллекта;
- анализ крови на уровень гормонов — позволяет выявить нарушение функций яичек и яичников;
- биохимический анализ крови — позволяет выявить признаки сопутствующего сахарного диабета в виде повышенного уровня глюкозы;
- ультразвуковое исследование внутренних органов — позволяет выявить сопутствующие анатомические аномалии их строения;
- осмотр офтальмолога — позволяет выявить снижение остроты зрения;
- электронейромиография — позволяет графически отобразить прохождение нервного сигнала по мышечным волокнам;
- молекулярно-генетическое исследование — является золотым стандартом и позволяет точно выявить дефект строения пятнадцатой хромосомы.

Выявление генетического дефекта — золотой стандарт диагностики синдрома Прадера — Вилли


Дифференциальный диагноз проводится со следующими заболеваниями, при которых основными признаками являются ожирение, низкий мышечный тонус, задержка интеллектуального развития:
- синдромом Дауна;
- миопатией;
- спинальной амиотрофией;
- синдромом Лоуренса — Муна;
- синдромом Барде — Бидля;
- синдромом Альстрема;
- синдромом Опитца — Фриаса.
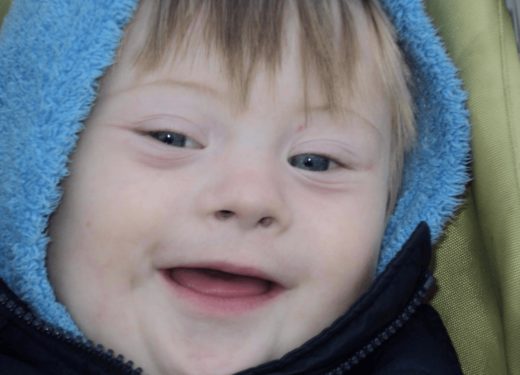
Наследственные синдромы — фотогалерея
-

- Болезнь Дауна — генетический дефект, связанный с изменением количества хромосом
-

- Больные с синдромом Лоуренса — Муна имеют характерный внешний вид

-

- Синдром Альстрема сопровождается развитием ожирения

Лечение наследственного ожирения
Лечение заболевания направлено на улучшение качества жизни больного, поскольку генетический дефект на современном этапе развития медицины исправить невозможно. Терапия при синдроме Прадера — Вилли комплексная, осуществляется коррекция питания и веса, мышечного тонуса, полового развития, а также исходных интеллектуальных способностей.
Медикаментозное лечение
Лекарственные препараты используются с целью достижения приемлемого уровня роста, а также правильного формирования половых органов в пубертатный период. Гормон соматотропин способствует росту мышц и скелета. Препарат используется до момента закрытия хрящевых зон длинных костей плеча, бедра, голени и предплечья.
Правильное половое развитие при синдроме Прадера — Вилли достигается назначением гормона гонадотропина. Препарат обеспечивает нормальный процесс формирования вторичных половых признаков.
Важно помнить, что только врач может решить, какова будет продолжительность курса лечения, назначить соответствующие препараты.
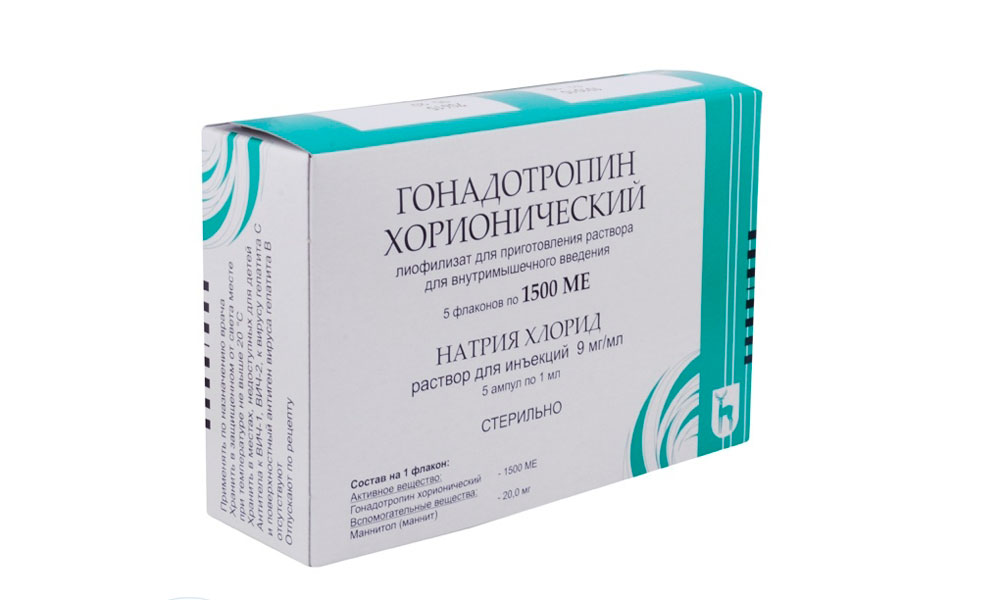
Для формирования вторичных половых признаков подросткам с синдромом Прадера — Вилли назначается Хорионический гонадотропин
Физиотерапия
Физиотерапевтические процедуры, используемые в раннем возрасте для коррекции мышечного тонуса:
- электростимуляция;
- электрофорез.
В основе этих методов лежит благотворное воздействие электрического тока различной формы и полярности. Массаж позволяет создать гармоничный тонус в различных группах мышц. Лечебная гимнастика — необходимое мероприятие при терапии больных с синдромом Прадера — Вилли. Наибольший эффект достигается при занятиях плаванием и аквааэробикой. Достаточный уровень физической активности жизненно необходим пациенту в борьбе с лишним весом.

Плавание — идеальный вид спорта для больного с синдромом Прадера — Вилли
Диета
Рациональное питание — основной способ коррекции особенного обмена жиров у больных. Прежде всего необходимо приучить ребёнка не переедать, поэтому родителям необходимо исключить свободный доступ к пище.
Рекомендуется к употреблению следующий набор продуктов:
- хлеб из цельных злаков;
- яркоокрашенные ягоды;
- свежевыжатые соки;
- кисломолочные продукты;
- морская рыба;
- морепродукты;
- зелёный чай;
- свежие овощи и фрукты;
- морская капуста.
Фотогалерея: продукты, рекомендуемые для включения в рацион
-

- Цельнозерновой хлеб — источник ценных углеводов

-

- Фруктовые натуральные соки содержат множество различных витаминов

-

- Морепродукты — источник ценного белка

-

- Зелёный чай содержит большое количество антиоксидантов

-

- Морская капуста — идеальный источник йода для поддержания функции щитовидной железы

-

- Кисломолочные продукты содержат кальций, необходимый для роста костей

Продукты, которых необходимо избегать:
- сахар;
- газированные напитки;
- фастфуд;
- жирные сорта мяса и рыбы;
- белый хлеб;
- картофель;
- шоколад;
- кондитерские изделия с кремом;
- свежая выпечка.
Фотогалерея: продукты, от которых следует отказаться
-

- Простые углеводы, содержащиеся в сахаре, ускоряют процессы отложения жира

-

- Фастфуд — не самая полезная и очень калорийная еда

-

- Белый хлеб из муки высшего сорта очень калориен

-

- Картофель — источник большого количество простых углеводов (крахмала)

-

- Кондитерские изделия отличаются повышенным содержанием сахара

-

- Газированные напитки содержат избыточное количество сахара и калорий

Коррекция интеллектуального развития ребёнка
Адекватный уровень интеллекта — залог социализации больных с синдромом Прадера — Вилли. Развитие исходных способностей на занятиях с логопедом-дефектологом и педагогом позволяет детям быть принятыми в любой коллектив.

Развитие правильного произношения и образной речи очень важно для ребёнка с синдромом Прадера — Вилли
Осложнения и прогноз заболевания
Своевременная постановка диагноза и адекватная терапия позволяет больным с синдромом Прадера — Вилли достичь приемлемого уровня качества жизни. При неблагоприятном течении заболевания развиваются следующие осложнения:
- сахарный диабет;
- тяжёлые формы остановки дыхания во время сна (апноэ);
- выраженное искривление позвоночника;
- разрушение хрящевой ткани суставов под влиянием избыточного веса;
- сердечная недостаточность;
- злокачественные новообразования.
При соблюдении всех врачебных рекомендаций продолжительность жизни больных с синдромом Прадера — Вилли может превышать шестьдесят лет.
Профилактика
Единственным эффективным методом профилактики является дородовая генетическая диагностика с определением особенностей хромосомного набора клеток плода, полученных из околоплодных вод. В последующем проводится консультирование врачом-генетиком.
Генетическое консультирование — единственный эффективный метод профилактики синдрома Прадера — Вилли
Синдром Прадера — Вилли — серьёзное генетическое заболевание. Для сохранения должного качества жизни необходимо длительное упорное сотрудничество ребёнка, его родителей и медиков. Только дисциплинированное выполнение всех рекомендаций сделает пациента полноценным членом современного общества.


